MEAP: mobile element annotation pipeline
MEAP (mobile element annotation pipeline) is a user-friendly pipeline for step-by-step completing bioinformatics and comparative genomics analyses of sequenced mobile genetic elements. It tells users how to carry out sequence collection and primary sequence annotation, annotation of backbone and accessory module sequences, drawing of various gene organization diagrams, and data submission. Please see our previous publications 1-5 for the examples of presentation and interpretation of mobile genetic element annotation results.
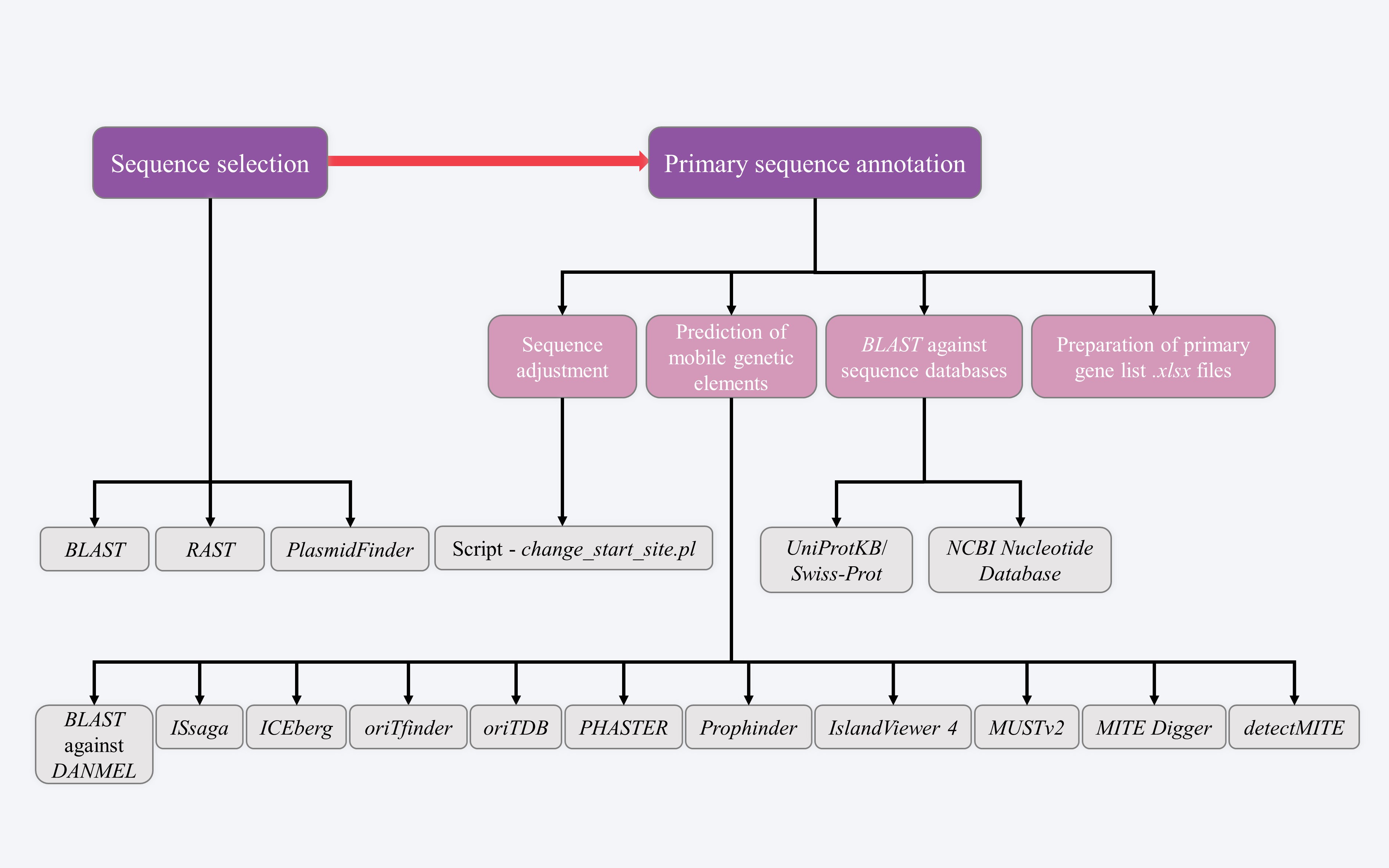
STEP 1 - Sequence collection and primary annotation
This step is to collect target sequences for analysis and further to perform primary sequence annotation.
1.1 Sequence selection
Users start with their own DNA sequence(s), e.g. the complete sequence of a plasmid, and then select one or more genetically related sequences from GenBank using BLAST; all these target sequences will be included in a subsequent bioinformatics and comparative genomics analysis. The primary automated sequence annotation is conducted using RAST, which generate the raw annotation data. The replicons of target sequences are identified using PlasmidFinder, or from RAST annotation results. The replicon is used to determine the Inc group of a plasmid.
1.1.1 BLAST
BLAST finds regions of similarity between sequences. The program compares nucleotide or protein sequences to various sequence databases (such as NCBI Protein Database, Refseq_genomes, Refseq_protein, Swissprot) and calculates the statistical significance.
1.1.2 RAST
RAST is a fully automated service for annotating complete or nearly complete bacterial and archaeal genomes. It provides high quality genome annotations for these genomes across the whole phylogenetic tree and can identify genomic features (i.e., protein-encoding genes and RNA) and annotating their functions.
Website: http://rast.nmpdr.org/
1.1.3 PlasmidFinder
PlasmidFinder identifies the Inc group of a plasmid in total or partial sequenced isolates of bacteria.
1.2 Primary sequence annotation
The primary annotation of target sequences contains four sections: sequence adjustment, prediction of mobile genetic elements, BLAST against sequence databases, and preparation of primary gene list files.
1.2.1 Sequence adjustment
The first base of start codon of repA (encoding replication initiation protein) of a plasmid or chromosome sequence, or that of IRi (inverted repeat at the integrase end) of an integron, or that of IRL (inverted repeat left) of a transposon is identified as the start site (+1) of a target sequence.
1.2.1.1 Script - Change start site
The Perl 5 script Change_start_site.pl changes the start site and also the + strand of a sequence in a FASTA file.
1.2.2 Prediction of mobile genetic elements
This step, using various softwares or databases, is to preliminarily predict mobile genetic elements (such as ISs, IS-based transposition units, composite transposons, MITE-flanking transposition units, MITE-like transposition units, unit transposons, ICEs, and IMEs) in each target sequence. Predicted mobile genetic elements will be validated and carefully annotated one by one in the following procedures.
1.2.2.1 BLAST against DANMEL
DANMEL is a database for annotation of mobile genetic elements associated with bacterial drug resistance.
1.2.2.2 ISsaga
ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes.
1.2.2.3 ICEberg
ICEberg is an integrated database that provides comprehensive information about integrative and ICEs found in bacteria.
Website: http://db-mml.sjtu.edu.cn/ICEberg/
1.2.2.4 oriTfinder
oriTfinder is a web-based tool that facilitates rapid identification of the origin site of DNA transfer (oriT) of a conjugative plasmid or a ICE or IME.
1.2.2.5 oriTDB
oriTDB is a web-based database, which currently contains details of transfer origin regions (oriT), nicking sites (nic), relaxases and auxiliary DNA-binding proteins, type IV coupling proteins, as well as a collection of directly related references.
1.2.2.6 PHASTER
PHASTER is a web server for the rapid identification and annotation of prophage sequences within bacterial genomes and plasmids.
Website: http://phaster.ca/
1.2.2.7 Prophinder
Prophinder is a tool dedicated to the detection of prophages in sequenced bacterial genomes based on BLASTP against the ACLAME database (http://aclame.ulb.ac.be/).
1.2.2.8 IslandViewer 4
IslandViewer is a computational tool that integrates four different genomic island prediction methods: IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander.
1.2.2.9 MUSTv2
MUSTv2 is a program for de novo discovering miniature inverted-repeat transposable elements from genomic sequences. It is implemented in Perl/C++ program languages, and BioPerl library is also used to process the sequence files.
1.2.2.10 MITE Digger
MITE Digger is a program for de novo discovering miniature inverted-repeat transposable elements from genomic sequences. It is implemented in Perl with Tk for graphic user interface for use on Microsoft Windows systems.
1.2.2.11 detectMITE
detectMITE is a tool based on MATLAB for detecting MITEs in genomes.
1.2.3 BLAST against sequence databases
All the protein or ORF (open reading frame) sequences will be BLASTed against NCBI Protein Database and UniProtKB/Swiss-Prot to clarify their name and function.
1.2.3.1 UniProtKB/Swiss-Prot
UniProtKB/Swiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB). It is a high quality annotated and non-redundant protein sequence database, which brings together experimental results, computed features and scientific conclusions.
1.2.3.2 NCBI Nucleotide Database
NCBI Nucleotide Database is a collection of nucleotide sequences from several sources, including GenBank, RefSeq, Third Party Annotation (TPA), and PDB.
1.2.4 Preparation of primary gene list .xlsx files
All raw annotation data of target sequences are collected into gene list .xlsx files, which will be extensively edited manually in subsequent procedures.
STEP 2 - Annotation of accessory modules
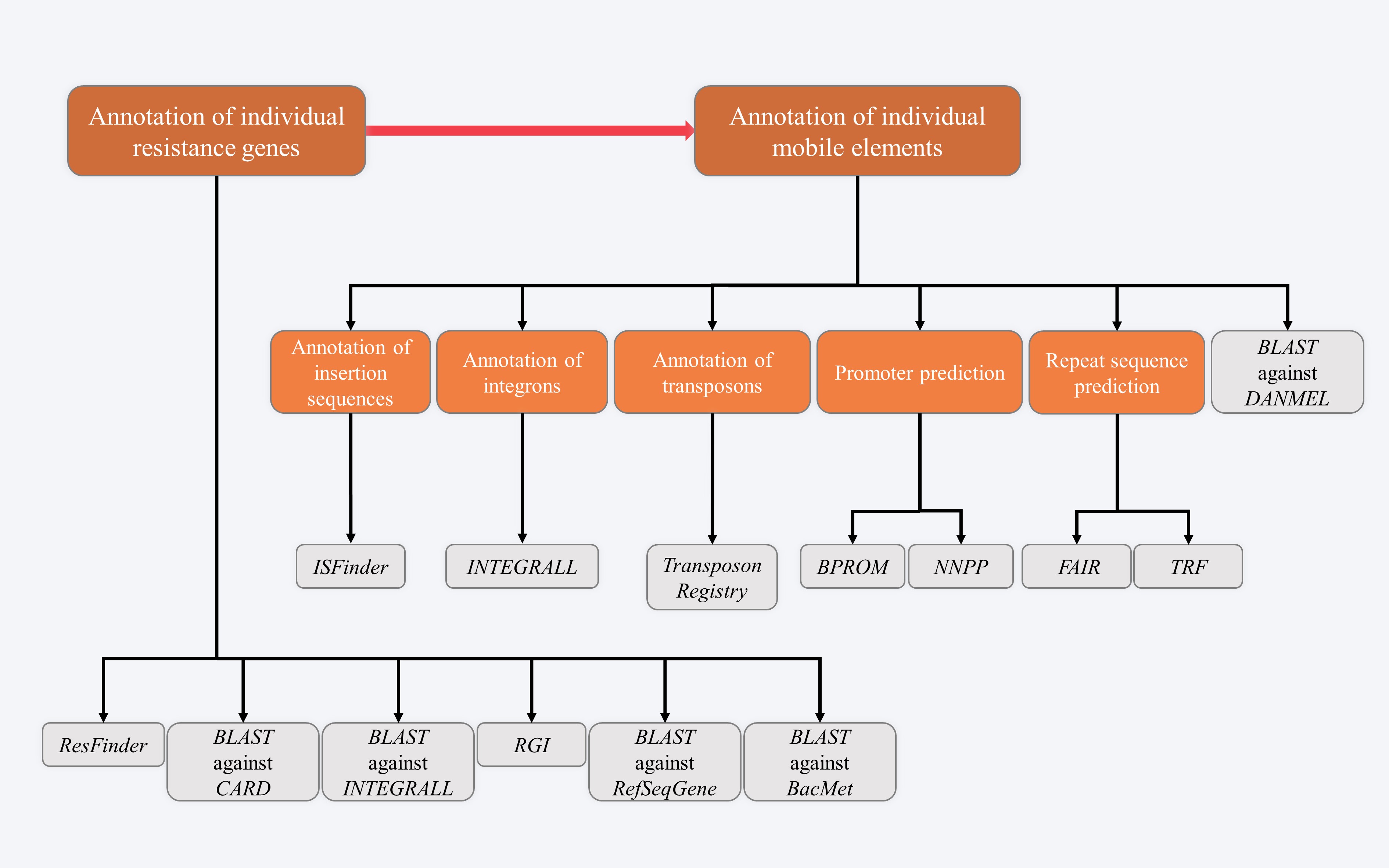
The modular structure of a target sequence (e.g. plasmid) is divided into the backbone sequences and the accessory modules (defined as acquired DNA regions associated and bordered with mobile genetic elements; inserted at different sites of the backbone). A deeper understanding of their diversification and evolution of mobile genetic elements is based on accurate annotation of their backbones and accessory modules. Although a number of softwares and databases can be used, extensive manual interpretation and editing are needed.
2.1 Annotation of individual resistance genes
First, submit target sequences to ResFinder to predict resistance genes and their resistance phenotypes. Second, verify resistance genes and their subtypes through various online databases, such as CARD, INTEGRALL, RGI, RefSeqGene and BacMet.
2.1.1 ResFinder
ResFinder is a web-based method, using BLAST for identification of acquired antimicrobial resistance genes and their resistance phenotypes in target sequences.
2.1.2 BLAST against CARD
CARD is a bioinformatic database of resistance genes, their products and associated phenotypes. Perform standard BLAST searches against the CARD reference sequences. Results are annotated with extra information from CARD.
2.1.3 BLAST against INTEGRALL
The INTEGRALL database is a freely available tool developed in order to provide an easy access to integron's DNA sequences and genetic arrangements. The INTEGRALL database only contains resistance genes carried by integrons.
Website: http://integrall.bio.ua.pt/?search
2.1.4 RGI
RGI in CARD provides a preliminary annotation of DNA or protein sequence(s).
Website: https://card.mcmaster.ca/analyze/rgi
2.1.5 BLAST against RefSeqGene
RefSeqGene, a subset of RefSeq project, defines genomic sequences to be used as reference standards for well-characterized genes.
2.1.6 BLAST against BacMet
BacMet provides a high quality, manually curated database of experimentally confirmed antibacterial biocide- and metal-resistance genes, and includes a database of predicted resistance genes.
Website: http://bacmet.biomedicine.gu.se/
2.2 Annotation of individual mobile genetic elements
Direct and inverted repeats, conserved genes or markers, intact/partial mobile genetic elements inserted, and resistance genes captured will be identified in each mobile genetic element.
2.2.1 Annotation of insertion sequences
2.2.1.1 ISFinder
ISFinder provides a list of insertion sequences isolated from bacteria and archae, and also contains the search and BLAST functions.
Website: https://www-is.biotoul.fr/index.php
2.2.2 Annotation of integrons
2.2.2.1 INTEGRALL
INTEGRALL is a freely available tool developed in order to provide an easy access to integron's DNA sequences and genetic arrangements.
Website: http://integrall.bio.ua.pt/
2.2.3 Annotation of transposons
2.2.3.1 Transposon Registry
Transposon Registry aims to simplify transposon nomenclature for new bacterial and archaeal elements and provide a searchable repository for all transposons to aid future research.
Website: http://transposon.lstmed.ac.uk/
2.2.4 Promoter prediction
2.2.4.1 BPROM
BPROM is a bacterial sigma70 promoter recognition program with about 80% accuracy and specificity. It is best used in regions immediately upstream from ORF start for improved gene and operon prediction in bacteria.
2.2.4.2 NNPP
NNPP is a method that finds eukaryotic and prokaryotic promoters in a DNA sequence.
2.2.5 Repeat sequence prediction
A typical integron or transposon is bordered by two inverted repeat sequences, which are needed for mobilization, and further bracketed by direct repeats (target site duplication signals) at both ends. The iterons are a tandem array of direct repeat sequences and located around repA, and they are RepA-binding sites essential for both initiation of plasmid replication and restriction of plasmid copy number.
2.2.5.1 FAIR
FAIR aims to finding all internal repeats in nucleotide sequence(s).
2.2.5.2 TRF
TRF is a program to locate and display tandem repeats in nucleotide sequence(s).
Website: http://tandem.bu.edu/trf/trf.html
2.2.6 BLAST against DANMEL
DANMEL is a database for analyzing mobile genetic elements associated with bacterial drug resistance.
STEP 3 - Annotation of backbone sequences
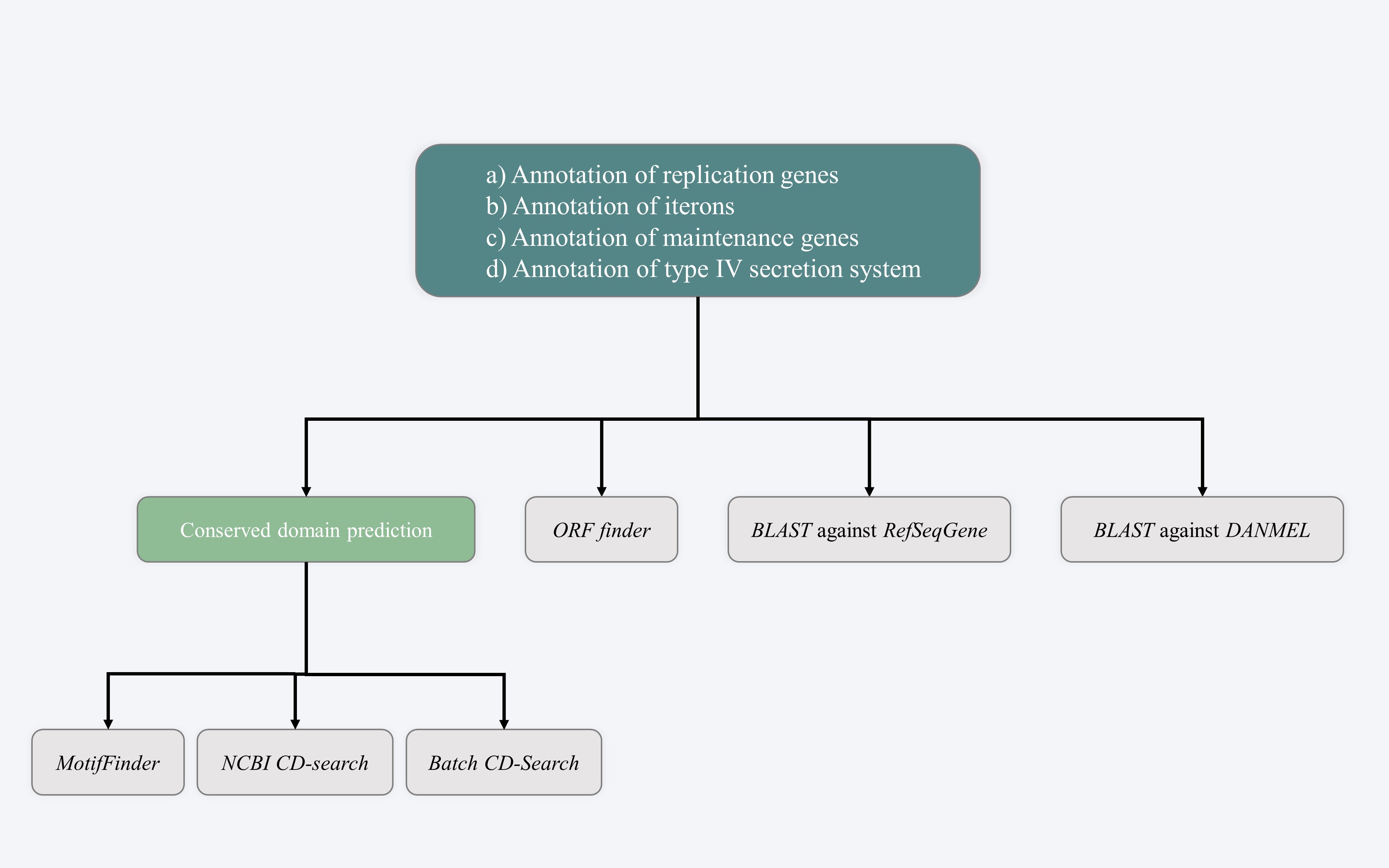
This step is to identify key genes/markers responsible for replication (e.g. replication genes and iterons), maintenance (e.g. partition, and toxin-antitoxin) and conjugal transfer (type IV secretion system) in target sequences (e.g. plasmids).
3.1 Conserved domain prediction
3.1.1 MotifFinder
MotifFinder finds out sequence motifs in the query sequence against Motif Libraries such as PROSITE, Pfam and NCBI-CDD, and provides functional and genomic information of the found motifs using DBGET and LinkDB as the hyperlinked annotations.
Website: http://www.genome.jp/tools/motif/
3.1.2 NCBI CD-search
CD-Search searches the Conserved Domain Database (NCBI-CDD; https://www.ncbi.nlm.nih.gov/cdd/) with protein or nucleotide query sequences. It uses RPS-BLAST to quick scan a set of pre-calculated position-specific scoring matrices with a protein query.
3.1.3 Batch CD-Search
Batch CD-Search serves as both a web application and a script interface for a conserved domain search on multiple protein sequences.
3.2 ORF finder
ORF finder searches for ORFs in the target DNA sequence.
3.3 BLAST against RefSeqGene
RefSeqGene, a subset of RefSeq project, defines genomic sequences to be used as reference standards for well-characterized genes.
3.4 BLAST against DANMEL
DANMEL is a database for analyzing mobile genetic elements associated with bacterial drug resistance.
STEP 4 - Drawing of gene organization diagrams
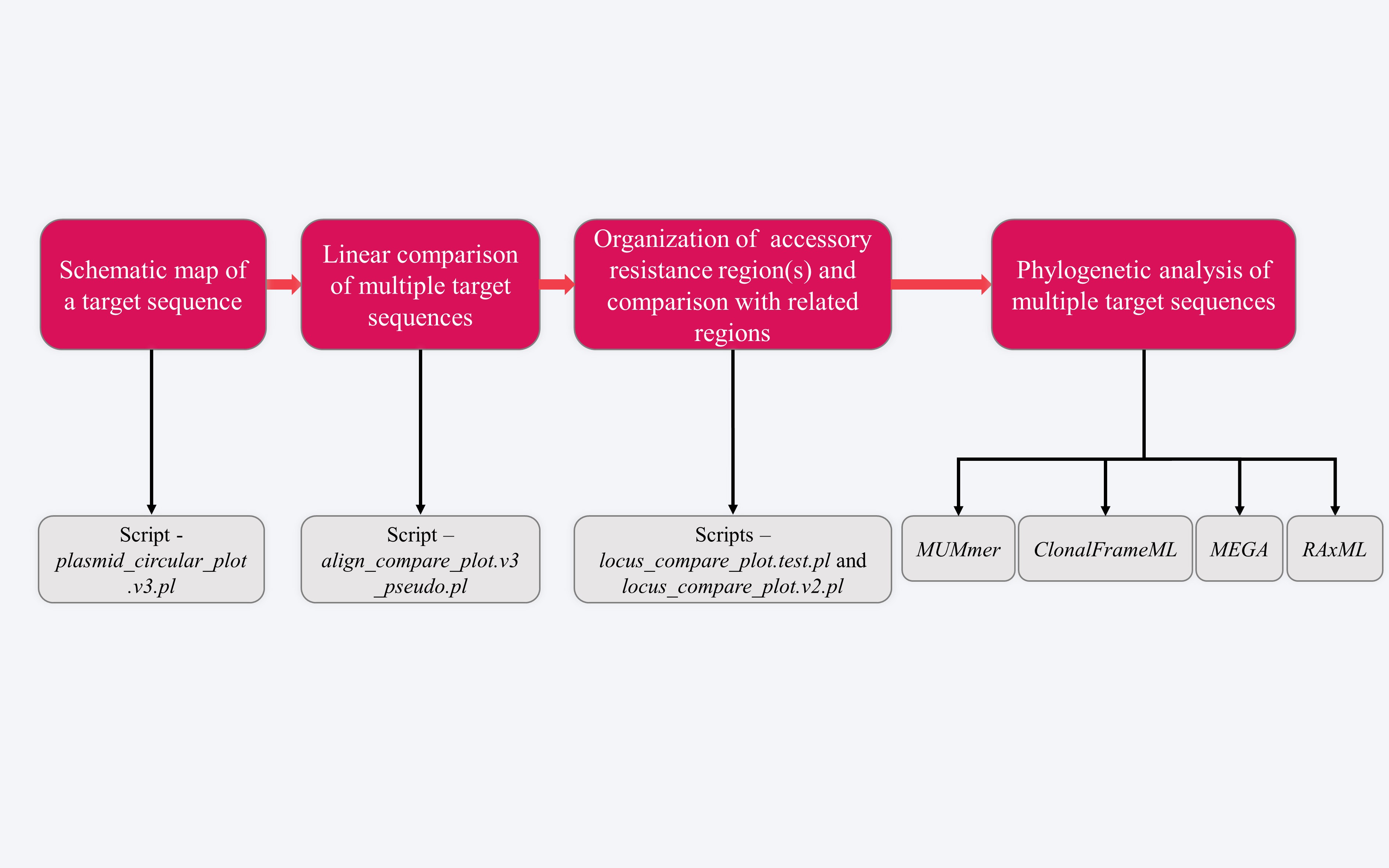
This step is to draw the schematic diagrams from the gene list .xlsx files of target sequences using Inkscape (https://inkscape.org), which is followed by extensive manual interpretation and editing. At least four figures will be presented: Figure 1 - Schematic map of a target sequence, Figure 2 - Linear comparison of multiple target sequences, Figure 3 - Organization of an accessory resistance region and comparison with related regions, and Figure 4 - Maximum-likelihood phylogenetic tree of multiple target sequences.
4.1 Schematic map of a target sequence
4.1.1 Script - plasmid_circular_plot.v3.pl
The custom Perl 5 script plasmid_circular_plot.v3.pl is used to draw the schematic circular map of a target sequence (e.g. a plasmid). Genes are denoted by arrows, and the backbone and accessory module regions are highlighted in black and color, respectively. The innermost circle presents GC-skew [(G-C)/(G+C)], with an indicated window size and an indicated step size. The next-to-innermost circle presents GC content.
4.2 Linear comparison of multiple target sequences
4.2.1 Script - align_compare_plot.v3_pseudo.pl
The custom Perl 5 script align_compare_plot.v3_pseudo.pl is used to draw the linear comparison of multiple target sequences. Genes are denoted by arrows. Genes, mobile genetic elements and other features are colored based on function classification. Shading denotes homology (with not less than an indicated nucleotide identity) of the sequences.
4.3 Organization of accessory resistance region(s) and comparison with related regions
4.3.1 Scripts - locus_compare_plot.test.pl and locus_compare_plot.v2.pl
The custom Perl 5 scripts locus_compare_plot.test.pl and locus_compare_plot.v2.pl are used to draw the organization of accessory resistance region(s) and comparison with related regions. Genes are denoted by arrows. Genes, mobile genetic elements and other features are colored based on their functional classification. Shading denotes regions of homology (with not less than an indicated nucleotide identity). Numbers in brackets indicate nucleotide positions within corresponding plasmids. These two scripts need to be used once each (with no order) to generate the diagram.
4.4 Phylogenetic analysis of multiple target sequences
In general, the backbone regions of indicative target sequences are aligned using MUMmer. Inference of homologous recombination is performed using ClonalFrameML to remove recombination-associated single-nucleotide polymorphisms (SNPs). A maximum-likelihood tree is constructed from recombination-free SNPs using MEGA or RAxML with a bootstrap iteration of 1000.
4.4.1 MUMmer
MUMmer is a tool for rapidly aligning entire genomes, whether in complete or draft form.
Website: http://mummer.sourceforge.net/
4.4.2 ClonalFrameML
ClonalFrameML is a software package that performs efficient inference of recombination in bacterial genomes.
4.4.3 MEGA
MEGA is an integrated tool for conducting automatic and manual sequence alignment, inferring phylogenetic trees, mining web-based databases, estimating rates of molecular evolution, and testing evolutionary hypotheses.
Website: https://www.megasoftware.net/
4.4.4 RAxML
RAxML is a program for sequential and parallel Maximum Likelihood based inference of large phylogenetic trees. It can also be used for post-analyses of sets of phylogenetic trees, analyses of alignments and evolutionary placement of short reads.
STEP 5 - Data submission
The gene list .xlsx file is checked using Sequin and then manually modified into a standard gene list .tab file, which is steadily recognized by BankIt. In addition, Sequin can be used to transfer the target sequence and its gene list .tab file into the standard .gbk and .sqn files. The target DNA sequence, together with its standard gene list .tab file and other relevant information, are submitted to GenBank using BankIt, to obtain an accession number.
5.1 Sequin
Sequin is a stand-alone software tool for submitting and updating sequences to the GenBank, EMBL, and DDBJ databases.
5.2 BankIt
BankIt is a web-based sequence tool for submitting sequences to GenBank.
References
1. Zhan Z, Hu L, Jiang X et al. Plasmid and chromosomal integration of four novel blaIMP-carrying transposons from Pseudomonas aeruginosa, Klebsiella pneumoniae and an Enterobacter sp. J Antimicrob Chemother 2018; 73: 3005-15.
2. Cheng Q, Jiang X, Xu Y et al. Type 1, 2, and 1/2-hybrid IncC plasmids from China. Front Microbiol 2019; 10: 2508.
3. Fang H, Feng J, Xu Y et al. Sequencing of pT5282-CTXM, p13190-KPC and p30860-NR, and comparative genomics analysis of IncX8 plasmids. Int J Antimicrob Agents 2018; 52: 210-7.
4. Jiang X, Yin Z, Yuan M et al. Plasmids of novel incompatibility group IncpRBL16 from Pseudomonas species. J Antimicrob Chemother 2020; 75 (8): 2093-100.
5. Liang Q, Jiang X, Hu L et al. Sequencing and genomic diversity analysis of IncHI5 plasmids. Front Microbiol 2018; 9: 3318.